Encapsulates many common "moves" you'll make when trying to make a heatmap, especially if you are trying to show geneset activity across a panel of samples.
NOTE: this function will almost certainly reorder the rows of the
input matrix. If you are concatentating Heatmap objects together horizontally
(ie. you if you want to use a rowAnnotation along side the returned heatmap),
you must reorder the rows of the annotation data.frame, ie.
ranno.df <- ranno.df[rownames(out@matrix),]
mgheatmap2(
x,
gdb = NULL,
col = NULL,
aggregate.by = c("none", "ewm", "ewz", "zscore"),
split = TRUE,
scores = NULL,
gs.order = NULL,
name = NULL,
rm.collection.prefix = TRUE,
rm.dups = FALSE,
recenter = FALSE,
rescale = FALSE,
center = FALSE,
scale = FALSE,
uncenter = FALSE,
unscale = FALSE,
rename.rows = NULL,
zlim = NULL,
transpose = FALSE,
...
)Arguments
- x
the data matrix
- gdb
GeneSetDbobject that holds the genesets to plot. Defaults toNULL, which will plot all rows inx.- col
a colorRamp(2) function
- aggregate.by
the method used to generate single-sample geneset scores. Default is
nonewhich plots heatmap at the gene level- split
introduce row-segmentation based on genesets or collections? Defaults is
TRUEwhich will create split heatmaps based on collection ifaggregate.by != 'none', or based on gene sets ifaggregate.by == "none".- scores
If
aggregate.by != "none"you can pass in a precomuptedscoreSingleSamples()result, otherwise one will be computed internally. Note that if this is adata.frameof pre-computed scores, thegdbis largely irrelevant (but still required).- gs.order
This is experimental, and is here to help order the order of the genesets (or genesets collection) in a different way than the default. By default,
gs.order = NULLand genesets are enumerated in alphabetical in the heatmap. You can pass in a character vector that will dictate the order of the genesets displayed in the heatmap. Currently this only matches against the"name"value of the geneset and probably only works whensplit = TRUE. We will supportcolleciton,nametuples soon. This can be a superset of the names found ingdb. As of ComplexHeatmap v2 (maybe earlier versions), this doesn't really work whencluster_rows = TRUE.- name
passed down to
ComplexHeatmap::Heatmap()- rm.collection.prefix
When
TRUE(default), removes the collection name from the genesets annotated on the heatmap.- rm.dups
if
aggregate.by == 'none', do we remove genes that appear in more than one geneset? Defaults toFALSE- recenter
do you want to mean center the rows of the heatmap matrix prior to calling
ComplexHeatmap::Heatmap()? This is passed down toscale_rows(). Look there for more mojo.- rescale
do you want to standardize the row variance to one on the values of the heatmap matrix prior to calling
ComplexHeatmap::Heatmap()? This is passed down toscale_rows(). Look there for more mojo.- center, scale, uncenter, unscale
boolean parameters passed down into the the single sample gene set scoring methods defined by
aggregate.by- rename.rows
defaults to
NULL, which induces no action. Specifying a paramter here assumes you want to rename the rows of the heatmap. Please refer to the "Renaming Rows" section for details.- zlim
A
length(zlim) == 2numeric vector that defines the min and max values fromxfor thecirclize::colorRamp2call. If the heatmap that is being drawn is "0-centered"-ish, then this defines the real values of the fenceposts. If not, then these define the quantiles to trim off the top or bottom.- transpose
Flip display so that rows are columns. Default is
FALSE.- ...
parameters to send down to
scoreSingleSamples(),ComplexHeatmap::Heatmap(),renameRows()internalas_matrix().
Value
A Heatmap object.
Details
More info here.
Renaming Heatmap Rows
This function leverages renameRows() so that you can better customize the
output of your heatmaps by tweaking its rownames.
If you are plotting a gene-level heatmap (ie. aggregate.by == "none"``) and the rownames()are gene identifiers, but you want the rownames of the heatmap to be gene symbols. You can perform this renaming using therename.rows` parameter.
If
rename.rowsisNULL, then nothing is done.If
rename.rowsis astring, then we assume thatxhas an associated metadatadata.frameover its rows and thatrename.rowsnames one of its columns, ie.DGEList$genes[[rename.rows]]orfData(ExpressionSet)[[rename.rows]]. The values in that column will be swapped out forx's rownamesIf
rename.rowsis a two-column data.frame, the first column is assumed to berownames(x)and the second is what you want to rename it to.When there are duplicates in the renamed rownames, the
rename.duplicates...parameter dictates the behavior. This will happen, for instance, if you are trying to rename the rows of an affy matrix to gene symbols, where we have multiple probe ids for one gene. Whenrename.duplicatesis set to"original", one of the rows will get the new name, and the remaning duplicate rows will keep the rownames they came in with. When set to"make.unique", the new names will contain*.1,*.2, etc. suffixes, as you would get from usingbase::make.unique().
Maybe you are aggregating the expression scores into geneset scores, and
you don't want the rownames of the heatmap to be collection;;name (or just
name when rm.collection.prefx = TRUE), you can pass in a two column
data.frame, where the first column is collection;name and the second
is the name you want to rename that to. There is an example of this in
the "Examples" section here.
Examples
# \donttest{
vm <- exampleExpressionSet()
gdb <- exampleGeneSetDb()
col.anno <- ComplexHeatmap::HeatmapAnnotation(
df = vm$targets[, c("Cancer_Status", "PAM50subtype")],
col = list(
Cancer_Status = c(normal = "grey", tumor = "red"),
PAM50subtype = c(Basal = "purple", Her2 = "green", LumA = "orange")))
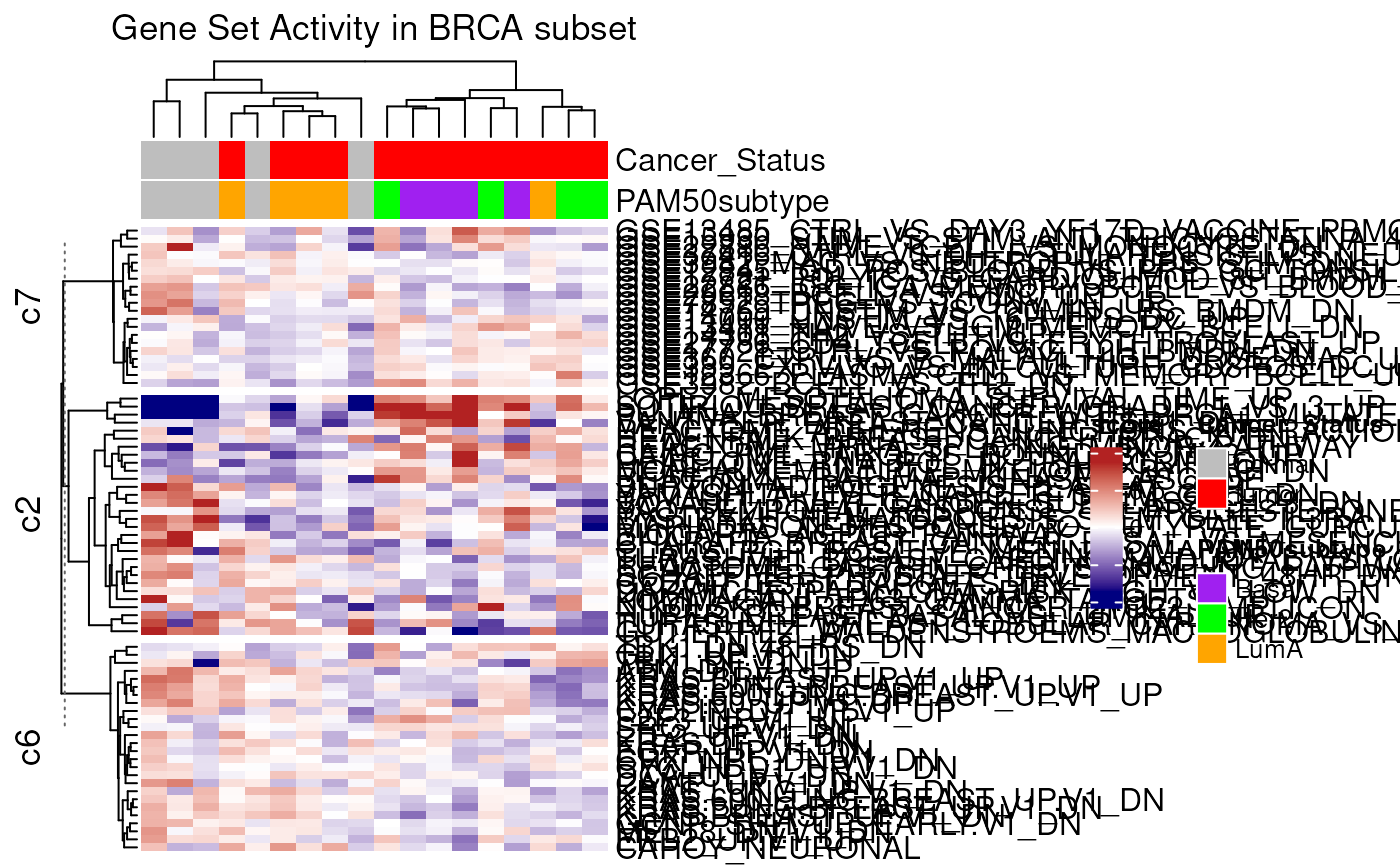
mgh <- mgheatmap2(vm, gdb, aggregate.by = "ewm", split = TRUE,
top_annotation = col.anno, show_column_names = FALSE,
column_title = "Gene Set Activity in BRCA subset")
ComplexHeatmap::draw(mgh)
 # Center to "normal" group
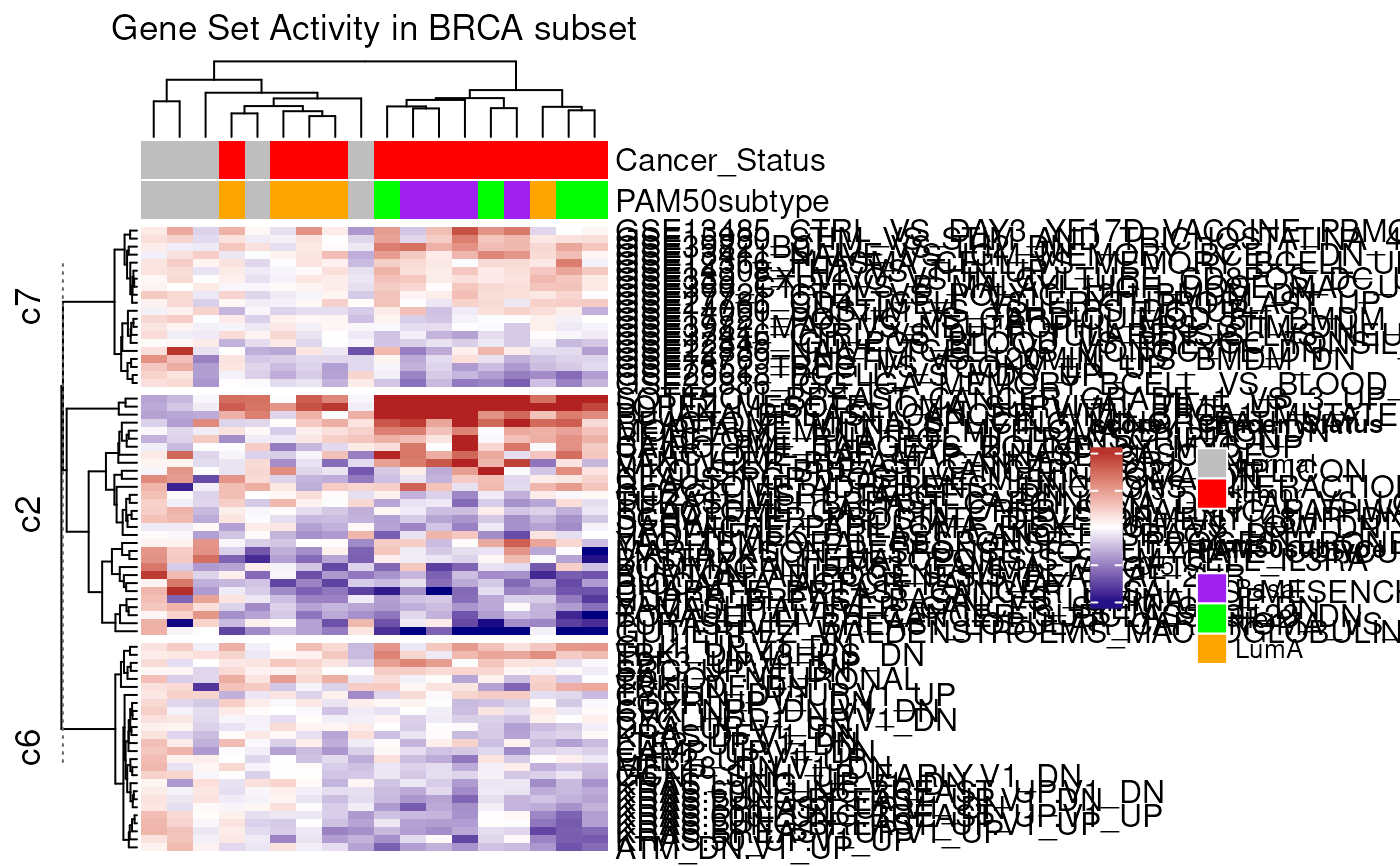
mgc <- mgheatmap2(vm, gdb, aggregate.by = "ewm", split = TRUE,
top_annotation = col.anno, show_column_names = FALSE,
recenter = vm$targets$Cancer_Status == "normal",
column_title = "Gene Set Activity in BRCA subset")
ComplexHeatmap::draw(mgc)
# Center to "normal" group
mgc <- mgheatmap2(vm, gdb, aggregate.by = "ewm", split = TRUE,
top_annotation = col.anno, show_column_names = FALSE,
recenter = vm$targets$Cancer_Status == "normal",
column_title = "Gene Set Activity in BRCA subset")
ComplexHeatmap::draw(mgc)
 # Maybe you want the rownames of the matrix to use spaces instead of "_"
rr <- geneSets(gdb)[, "name", drop = FALSE]
rr$newname <- gsub("_", " ", rr$name)
mg2 <- mgheatmap2(vm, gdb, aggregate.by='ewm', split=TRUE,
top_annotation = col.anno, show_column_names = FALSE,
column_title = "Gene Set Activity in BRCA subset",
rename.rows = rr)
# }
# Maybe you want the rownames of the matrix to use spaces instead of "_"
rr <- geneSets(gdb)[, "name", drop = FALSE]
rr$newname <- gsub("_", " ", rr$name)
mg2 <- mgheatmap2(vm, gdb, aggregate.by='ewm', split=TRUE,
top_annotation = col.anno, show_column_names = FALSE,
column_title = "Gene Set Activity in BRCA subset",
rename.rows = rr)
# }